Research |
NGS facility |
Publications |
Laboratory |
People |
Photos |
Science |
Research Interests |
The research in our laboratory is focused on the studies of complex microbial communities associated with human gastrointestinal system. We use a variety of research techniques including ribosomal gene sequencing, metagenomics, phylogenetic microarrays, and fluorescent in situ hybridization to gain knowledge of community composition and function, its changes in disease, and its response to diet perturbations. We also associate microbial dynamics to changes in luminal and fecal metabolites in the same samples. We employ mathematical modeling to generate hypotheses of possible microbial and host-microbial interactions, that we then test in the in vitro human gut simulator system. We also study individual microbial species to gain insight into the specific roles these members play in our lives and how they interact with each other. Below we describe our recent research projects in more detail, and also list experimental and computational approaches that we use. |
|
|
Typical human body contains about 1012-1013 eukaryotic cells. However, most of the cells in our body are not our own, nor are they even human. They are microbial. From the invisible strands of fungi waiting to sprout between our toes to the kilogram of bacterial matter in our guts, humans house about 1013-1014 microbial cells. Almost every epithelial surface of our body contains a complex microbial community – skin, mouth, vagina, and gastrointestinal tract. Microbes can live even in the highly acidic environment of the stomach (pH=2). All these epithelial surfaces are populated by communities of microorganisms, collectively called microbiota, rather than by individual species, and often include hundreds of different microbial members. Majority of these organisms are bacteria, though archaea, fungi, eukaryotic microorganisms, and viruses also take residence in different epithelial niches. |
|
We now begin to understand the important relationships these microbes form with our bodies. For example, microbiota of the gut participate in our energy metabolism by breaking down complex polysaccharides in the diet. Resident microbes also protect us from pathogen invasion through competition for resources, or directly by inhibiting pathogen growth. Moreover, microbiota modulate the proper development and functioning of the human immune system, transform or excrete toxic substances, and help maintain gut well-being. At the same time, microbiota dysbiosis, defined as the perturbation of the normal microbial profile, has been linked to a number of human diseases including dental plaque, bacterial vaginosis, psoriasis, atopic dermatitis, and cystic fibrosis. The focus of our laboratory is on the analysis of microbes colonizing human gastrointestinal system. In each healthy human, this gut community contains several hundreds different species, which display complex network of interactions not only among themselves but also with the human tissues and cells. Many recent studies from our and other groups also showed that alterations in the levels of certain microbial groups in the gut may be linked to several disorders including irritable bowel syndrome, inflammatory bowel disease, obesity, diabetes, and colon cancer. |

Distribution and abundance of bacteria in human gastrointestinal tract. [Figure modified from B. Sartor Gastroenterology 2008] |
Different approaches can be taken to study these complex microbial populations. We can look at the community membership where interrogation of small ribosomal subunit (SSU) RNA genes is used to obtain the phylogenetic structure of the community. We can profile community functionality by cataloguing the pool of functional genes and proteins present in the population through meta-genomic, meta-transcriptomic, and meta-proteomic approaches. We can also study the metabolic processes in microbial conglomerates by measuring the levels of produced and consumed metabolites . We can employ in vitro and animal model systems to carry out controlled studies of possible metabolic and immunological links among microbes and with host cells. And we can use a variety of computational and bioinformatic tools to link these diferent types of data together to derive significant associations and possible cause-effect hypotheses. The goal of such research is to learn sufficient detail about human-associated microbiota to allow basic scientists and medical doctors to design new options to prevent, diagnose, and treat many of the diseases mentioned above. Microbial community profiling can be used in clinical setting to diagnose many different diseases non-invasively and relatively inexpensively. Live microbial species with discovered beneficial properties called probiotics can be prescribed to patients in order to influence microbiota composition and shift this community back into a healthier state. Specific mixtures of probiotic species can be provided to each individual patient based on the particular disbalanced state of their microbiota (personalized medicine). Finally, a complete replacement of patient microbiota can also be carried out to treat severe cases of microbiota disbiosis, as is the case of the use of fecal microbiota transplantation to cure Clostridium difficile bacterial infection of the gut. |
|
 Introduction to human intestinal microbiota
Introduction to human intestinal microbiota
|
For an up to date list of publications, please visit the Publications page. |
|
How do different diet supplementations affect human gut microbiota? [S. Rajakaruna et al Microorganisms 2022; Perez-Burillo et al Food Chem. 2020, Food Sci. Technol. 2019, and Food Chem. 2019; Agans et al Tox Sci. 2019] |
|
|
Several recent projects in our laboratory have investigated how different food additives can affect human gut microbes. To specifically elucidate the action of these compounds on the gut microbiota (and not secondary effect from host response), in several of these projects we used the Human Gut Simulator, an in vitro system that we designed to mimic conditions of the human intestinal environment (see brief description of the system below in techniques section). First, we studied how human gut microbes ferment green and roasted coffees [Perez-Burillo et al Food Chem. 2019]. Fermentation of coffee brews led to different community structure between green and roasted coffees. Metabolism of green coffees generated more antioxidants and higher levels of short chain fatty acids (SCFAs, end-products of microbial metabolism beneficial to human health). This suggested that green and roasted coffees behave as different types of food. Next, in collaboration with meat producing company, we asked if nutritional qualities of salami sausage can be improved by health-promoting supplements [Perez-Burillo et al Food Sci. Technol. 2019]. The addition of different types of dietary fiber (citrus fiber, arabinogalactan, and inulin), a probiotic Lactobacillus rhamnosus, and an herbal extract to the salami formulation was tested. Incorporating dietary fiber into salami formulation increased sausage antioxidant capacity and the amount of SCFAs produced during microbiota fermentation. Citrus fiber and arabinogalactan but not inulin promoted an increase in the abundance of several known polysaccharide degrading genera and resulted in a reduction in the abundance of Escherichia, a bacterial genus known to contain human intestinal pathogens (see Figure). Overall, we found that the addition of dietary fiber to salami formulation prior to curing improved beneficial health markers of this food product. Because of ever increasing industrialization of our lives, people also come into contact with many chemicals such as different nanomaterials. We wanted to see how ingestion of different nanoparticles, namely titanium oxide and nanosilver can affect our gut microbes [Agans et al Tox. Sci. 2019]. Addition of TiO2 to microbial communities led to a modest reduction in community density but had no impact on community diversity and evenness. In contrast, administration of known antimicrobial silver nanoparticles resulted in a drastic reduction of population density. In both cases, communities recovered once the addition of nanomaterials was ceased. Finally, we also wanted to unravel whether gut microbes can degrade melanoidins, an abundant component of many foods (when you brown a toast, that color is caused by the formation of such melanoidins) [Perez-Burillo et al Food Chem. 2019]. In our study, we subjected purified melanoidins from a variety of foods (coffee, bread, beer, balsamic vinegar, sweet wine, biscuit, chocolate, and breakfast cereals) to digestion and fermentation by human gut microbiota. Some melanoidins were extensively used by gut microbes, increasing production of short chain fatty acids (mainly acetate and lactate) and favoring growth of the beneficial genera Bifidobacterium (bread crust, pilsner and black beers, chocolate and sweet wine melanoidins) and Faecalibacterium (biscuit melanoidins) (see Figure). Gut microbes were also able to release phenolic compounds initially linked to the melanoidin backbone. Melanoidins can thus be considered as potential prebiotic agents. |
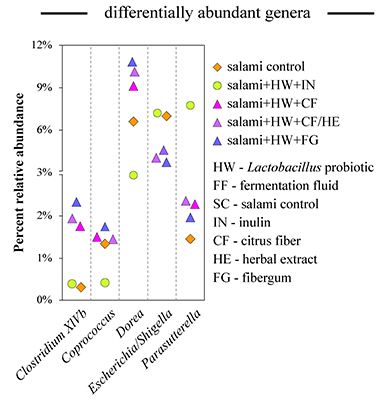 Figure. Fermentation of salami preparations with fiber by human fecal microbiota increases prevalence of different bacterial genera. [Perez-Burillo et al Food Sci. Technol. 2019] 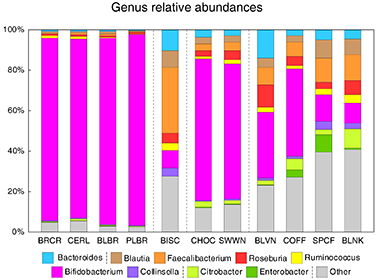 Figure. Fermentation of melanoidin preparations by human fecal microbiota promotes different community structures. Each column represents a community derived from a fermentation of particular melanoidin. BRCR – bread crust; CERL – cereal; BLBR – black beer; PLBR – pilsner beer; BISC – biscuits; CHOC – chocolate; SWWN – sweet wine; BLVN – balsamic vinegar; COFF – coffee; SPCF – spent coffee; BLNK – blank. [Perez-Burillo et al Food Chem. 2020] |
|
Dietary fatty acids sustain the growth of the human gut microbiota. [Agans et al Appl. Environ. Microbiol. 2018] Increased intake of fats in many developed countries raised awareness of potentially harmful and beneficial effects of high fat consumption on human health. Some dietary fats escape digestion in the small intestine and reach the colon. Many dietary effects on human health are mediated through the function of gut microbes. While we understand sufficiently well how gut microbes ferment carbohydrates and fats, much less is known about microbial degradation of dietary fats in the human gut. |
|
|
In this study we used an in vitro multi-vessel simulator system of the human colon (HGS) to test if human gut microbes can sustain their population when the dietary fats are the only readily available sources of carbon in the environment. We first established a stable community in each HGS vessel on a balanced medium that resembled colon influent on a typical Western diet. This Western medium (WM) contained complex and simple saccharides, proteins, and fats in proportions matching previously reported amounts in the human colon. After 14 days of stabilization, the WM medium was switched to a “fats-only” variant (FOM), where all carbohydrates and proteins were removed, with the exception of a small amount of yeast extract. Microbiota of the proximal vessel responded to this change in the supplied medium within 24 hours as was evident from the sharp drop in the community cell density on day 15 (see Figure on the right). Similar initial decline in the cell density was also observed in the transverse and distal vessels. Nevertheless, a dense population of microbes was maintained in all vessels on the FOM. We confirmed in a control run with yeast-only medium that dietary fats were indeed necesary to mintain this community density. The community composition on the balanced Western medium was predictably dominated by members of classes Clostridia and Bacteroidia. The switch to FOM led to a significant reduction in the overall abundance of Clostridia and Verrucomicrobiae, with a smaller decrease in Bacteroidia. In contrast, the numbers of different clades of Proteobacteria increased. The overall production of short-chain fatty acids, the main fermentation end-products in the gut, was reduced dramatically upon removal of carbohydrates and proteins from the medium. Acetate production was impacted the least, likely because it is a direct end-product of the β-oxidation of fatty acids. Indeed, at the functional level we found that the abundances of most enzymes of the fatty acid degradation pathway were higher in the communities maintained on the fats-only medium (see Figure below). We thus showed that human gut microbes are able to maintain a complex community when supplied with dietary fatty acids as the only nutrient and carbon sources. Such fatty acid based growth leads to lower production of short chain fatty acids and antioxidants by community members, which might potentially have negative health consequences on the host. |
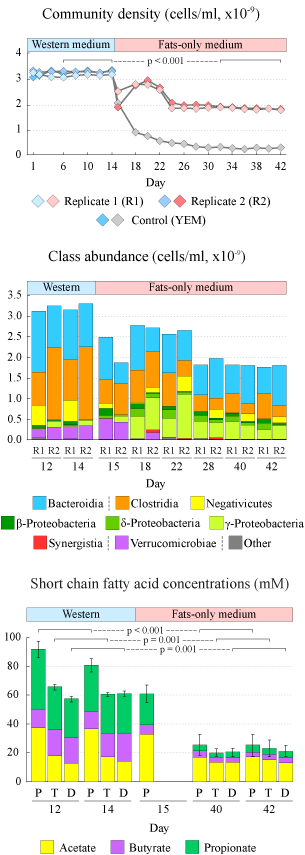 Figure. Dynamic changes in community density (top panel), absolute class baundances (middle panel), and produced short chain fatty acids (bottom panel). |
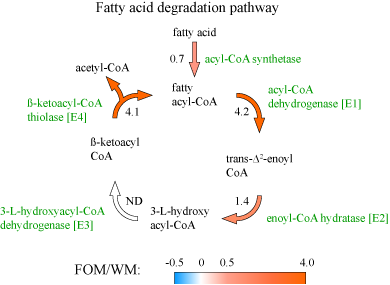 Figure. The predicted log2-transformed abundance ratios (fat-only medium/Western medium) for the genes encoding the enzymes of fatty acid β-oxidation pathway. |
|
|
Differences in gut metabolites and microbial composition and functions between Egyptian and US teenagers are consistent with consumed diets. [Shankar et al mSystems 2017] |
|
|
Throughout human evolution dramatic shifts in lifestyle and geographical distribution of the human species led to periodic changes in consumed diets and nutritional intakes. These shifts in dietary habits have historically given rise to several different diet types. Typical “Western” diet consumed by the majority of populations in most industrialized countries is rich in animal proteins and fats, dairy products, and refined, starch-enriched grains, cereals, flour, and sugars. In contrast, the “Mediterranean” diet is high in fruits, vegetables, whole grains, beans, nuts, and plant fats, whereas the fraction of meats and sweets is low. Populations consuming Mediterranean diet have significantly lower incidence of cardiovascular and metabolic diseases compared to industrialized societies, and it is important to determine the reasons for such improved health. Human gastrointestinal microbiota functions as an important mediator of diet on host metabolism. Many different compounds, primarily fiber and other polysaccharides, but also some proteins, fats, and simple sugars reach colon where they all are fermented by resident gut microbes. In this study we aimed to discover possible relationships between human gut microbiota and consumed diets. To achieve that goal, we compared fecal microbiota structure and function as well as fecal metabolites in two cohorts of children: teenagers from the United States consuming a typical Western diet, and Egyptian teenagers consuming a Mediterranean type diet. We found that the gut microbiota in each population has adapted to the host’s prevalent diet. Thus, Egyptian gut microbiota was enriched in polysaccharide degrading enzymes and in microbial members well suited to fiber degradation. The microbial communities in US teenagers had higher counts of protein-degrading microbes and were enriched in the protein and fat-utilization pathways as well as in the biosynthesis of vitamins that are often low in Western diets. Such differences in microbiota structure and function were reflected in the differences in the intestinal metabolites. While the gut environment of Egyptian teenagers was characterized by abundance of short chain fatty acids (end products of fiber degradation), intestines of US children had increased amino acid content, higher levels of lipid metabolism associated compounds, and elevated concentration of 1-methylhistamine, a biomarker for allergic reactions. Short chain fatty acids, especially butyrate, inhibit inflammation and protect against obesity, whereas products of protein and lipid degradation are associated with a risk of developing atherosclerosis and colon cancer. Overall, we can hypothesize that intestinal microbiota might be selected based on diets we consume, which can open opportunities to affect gut health through gut microbiota modulation with dietary supplementations such as pre- and probiotics. |
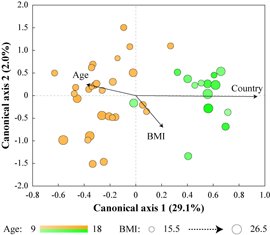 Figure. Separation of Egyptian (yellow) and US (green) samples based on their microbiota composition. Influence of country, age and BMI on the distribution of samples is shown by size and direction of arrows. 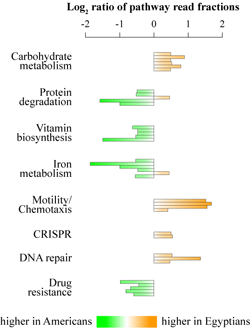 Figure. Differential abundances of select mcirobial functions. |
|
Simultaneous interrogation of fecal microbiota and metabolite profiles reveals differences in the microbe-metabolite associations between healthy and IBS-D children. [Shankar et al ISME 2015] Many pathophysiological effects in the gut environment are the result of complex metabolic interactions between gastrointestinal microbiota and the host. Microbial degradation and fermentation of host dietary compounds lead to production of end-products that can have profound impact of the host health. Thus, valuable insights into links between gut microbiota interactions and GI diseases can be obtained by simultaneous interrogation of microbes and metabolites of the gut environment. With such goal we extracted aqueous metabolites from 44 fecal samples obtained from healthy adolescent children and adolescents diagnosed with diarrhea-predominant irritable bowel syndrome (IBS-D), and we utilized proton nuclear magnetic resonance for high-throughput metabolomics analysis. The metabolite profiles were compared to the microbiota composition profiles obtained from the same set of samples. Spearman rank correlation analysis revealed many strong microbe-metabolite associations in the healthy sample set (see Figure below), while no such associations were observed in the IBS set. This observation mimics our previous report where gut microbe-microbe interactions were found to be severely limited in IBS-D. In addition, by utilizing several different multivariate statistical techniques, we showed that healthy and IBS samples could be distinguished based on fecal metabolite profiles, and such separation was similar to the microbiota based separation. Measurements of individual metabolites indicated that the intestinal environment in IBS-D was characterized by increased proteolysis, incomplete anaerobic fermentation and possible change in methane production. These findings show that fecal microbial and metabolite profiling can be used to facilitate IBS-D diagnosis. |
|
 Figure. Clustering of Spearman rank correlation values between microbial genus abundances and metabolites in fecal samples from healthy adolescents. Yellow boxes highlight statistically significant correlations. Select significant metabolite-microbial assocaitions are visulaized on the right image panel. |
|
|
Analysis of gut microbiota in Clostridium difficile infected patients following fecal microbiota transplantation. [Shankar et al Microbiome 2014] Clostridium difficile is an opportunistic human intestinal pathogenic bacterium, and C. difficile infection (CDI) is one of the main causes of antibiotic-induced diarrhea and colitis that can lead to death. One successful approach to combat CDI, particularly recurrent form of CDI, is through transplantation of fecal microbiota from a healthy donor to the infected patient. In this technique, fecal microbiota obtained from a healthy donor is processed, standardized, and subsequently transplanted into patients suffering from the recurrent C. difficile infection. FMT is highly successful (>90% success rate). |
|
|
In this study we investigated the distal gut microbial communities of several CDI patients before and after fecal microbiota transplantation, and we compared these communities to the composition of the donor’s fecal microbiota. The timeline of the procedure and sample collection are shown in the image. We utilized phylogenetic Microbiota Array, high-throughput Illumina sequencing, and fluorescent in situ hybridization to profile microbiota composition down to the genus and species level resolution. The colon of each CDI patient was host to a severely compromised intestinal microbial community, which was significantly reduced in diversity and richness. This was likely a result of C. difficile proliferation as well as due to the antibiotic treatment used in an attempt to rid the disease. While the healthy gut microbiota is usually dominated by bacterial classes Clostridia and Bacteroidia, fecal samples of the CDI patients were abundant in Gammaproteobacteria and Bacilli. In comparison, the donor community was dominated by Clostridia and had significantly higher diversity. The patients' distal gut communities were completely transformed within three days following fecal transplantation. The transplanted gut communities were highly similar among recipients post-transplantation and were indistinguishable from that of the donor (see PCoA image). In each case, the gut microbiota restoration led to a complete patient recovery and symptom alleviation. The transplanted communities remained stable in each patient for at least four months. Thus, we showed that C. difficile infection can be successfully eradicated by fecal microbiota transplantation, which offers simple and highly effective (>90% success rate) treatment option. |
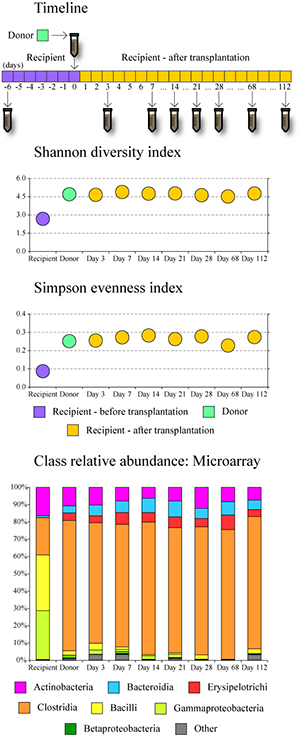 Figure. Changes in microbiota diversity and composition following fecal transplantation in CDI patients. |
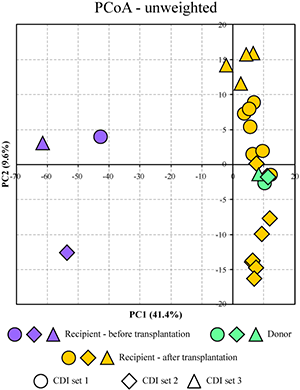 Figure. Separation of all profiled samples in PCoA space. |
|
|
Quantitative profiling of gut microbiota of children with diarrhea-predominant irritable bowel syndrome. [Rigsbee et al Amer J Gastroenterol 2012 and Shankar et al Gut Microbes 2013] |
|
|
The goal of this study was to determine the distal gut microbiota in two groups of pre- and adolescent children: healthy volunteers and children diagnosed with diarrhea-predominant irritable bowel syndrome (IBS-D). A focus on adolescent children was justified by the higher prevalence of IBS in this population demographic. Phylogenetic Microbiota Array was used to obtain quantitative measurements of bacterial presence and abundance in subjects’ fecal samples. We utilized high-throughput DNA sequencing, quantitative PCR, and fluorescent in situ hybridization to confirm microarray findings. Both sample groups were dominated by the phyla Firmicutes, Bacteroidetes, and Actinobacteria, which cumulatively constituted 91% of overall sample composition on average. A core microbiome shared among analyzed samples encompassed 55 bacterial phylotypes dominated by genus Ruminococcus; members of genera Clostridium, Faecalibacterium, Roseburia, Streptococcus, and Bacteroides were also present. The core was smaller for the IBS group, and most analyzed samples contained unique phylotypes only detected in that single sample (see image). Similarly to the previous project, by utilizing ordination and classification computational techniques such as PCA, PCoA, and OPLS-DA, we were able to determine sample origin (healthy or IBS) based on its microbiota composition. Several genera were found to be differentially abundant in the gut of healthy and IBS groups: levels of Veillonella, Prevotella, Lactobacillus, and Parasporobacterium were increased in children diagnosed with IBS, whereas members of Bifidobacterium and Verrucomicrobium were less abundant in those individuals. By calculating a nonparametric correlation matrix among abundances of different genera in all samples, we also examined potential associations among intestinal microbes. Strong positive correlations were found between abundances of Veillonella and both Haemophilus and Streptococcus, between Anaerovorax and Verrucomicrobium, and between Tannerella and Anaerophaga. Many positive associations were lost in IBS that can potentially indicate a loss of important mutualistic interactions among microbiota members in this disease - this can lead to changes in metabolite levels in intestinal lumen, which in turn can result in host physiological response. |
 Figure. Core microbiome in IBS (A) and healthy (B) children. 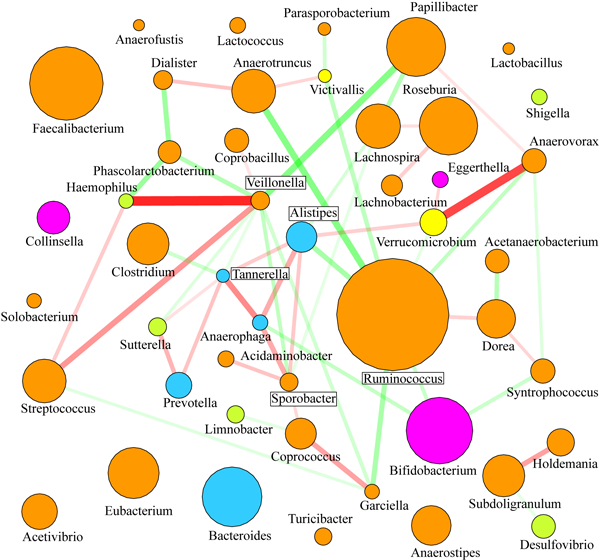 Figure. Associations among abundances of microbial genera [use "view image" to expand]. |
|
Analysis of distal gut microbiota in adolescent children and adults. [Agans et al FEMS-ME 2011] Several projects in our laboratory have looked at the gut microbiota in teenage children. In this work, we used phylogenetic Microbiota Array, developed previously in our laboratory, for high-throughput analysis of distal gut microbiota in healthy adolescent children 11-18 years of age. Samples obtained from healthy adults were used for comparison. We were able to separate child and adult samples based solely on the composition of their fecal microbiota (principal components analysis, see figure). All samples were dominated by class Clostridia. While we have detected many different genera in analyzed samples, the top 12 abundant genera contributed three-forth of total microbial abundance. All the other genera had relatively low representation. Genus Ruminococcus, which contains many prominent polysaccharide degrading members, was by far the most abundant. Genera Bifidobacterium and Clostridium were more abundant among adolescent samples than those from adults. Overall, this study suggested that the gut microbiome of adolescent children is different from that of adults. |
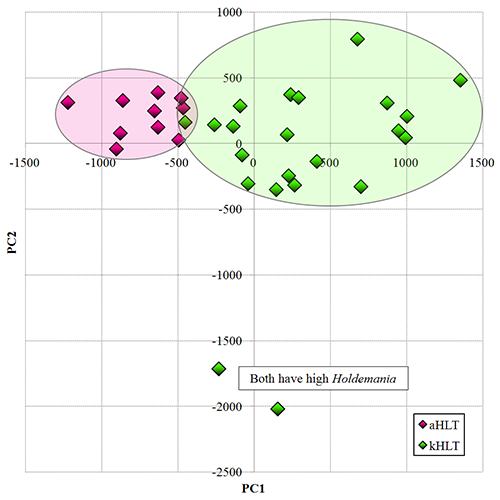 Figure. Separation of adolescent (kHLT, green) and adult (aHLT, purple) samples in PCA space. |
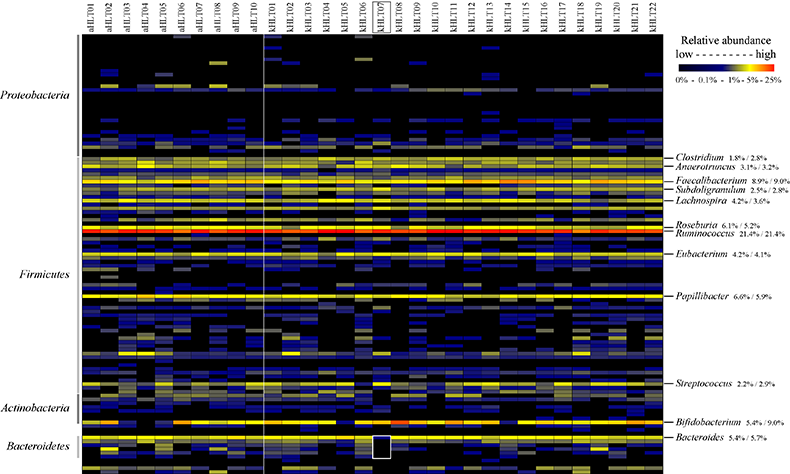 Figure. Distribution of genus abundances among profiled adult and adolescent samples. |
|
|
|
Clinical studies. We work closely with a number of medical doctors and hospitals with an aim to profile human gut microbial communities and determine potential differences in these communities in patients with different GI disorders. Such studies can be done cross-sectionally where we identify a group of people all diagnosed with a particular disorder, and then obtain either a stool or a biopsy sample from them at one specific time. Alternatively, a longitudinal analysis can be performed, where each patient, which enrolled in a study, provides a number of samples over a period of time. Whereas the first approach allows us to compare microbiota differences between two different gut states (usually healthy vs disease), the second approach lets us follow microbiota dynamic changes in the patients' GI tracts as they receive treatment or go through different procedures. |
 |
To date we have profiled gut microbiota in patients diagnosed with irritable bowel syndrome (IBS), inflammatory bowel disease (IBD) and Clostridium difficile infection. We have also compared gut microbial communities in different populations of healthy people - children and adults, healthy adults undergoing specific diet changes, and children receiving probiotic and prebiotic supplementations. |
|
In vitro human gut simulator. To complement our clinical studies we have established and validated an in vitro simulator of human intestinal tract (see image below). The simulator consists of three continuously linked fermentation vessels, each set to mimic environmental conditions of different sections of human GI tract known to house the majority of gut microbes: vessel 1 simulates the ascending colon, vessel 2 simulates the transverse colon, and vessel 3 models the descending colon. The medium, which closely matches food bolus contents that reach colon, is supplied to vessel 1, and vessel contents are periodically transferred from vessel 1 to vessel 2, from vessel 2 to vessel 3, and from vessel 3 to waste collection flask. Additional vessels simulating stomach and small intestinal regions of the GI tract can also be added when needed. The environmental conditions (temperature, pH, relative volume, peristaltic movement, dilution with lumenal contents, etc) in each vessel closely match those experimentally measured in the corresponding sections of the human gut. This bioreactor is able to maintain high microbial density over weeks of continuous incubation in conditions that can be precisely controlled by an investigator. Because we can change environmental conditions in this system at will, can draw samples at any point, and can precisely perturb the community, this system is well suited to carry out human gut microbiota studies that so far have not been done in the field. We can obtain samples from each simulated gut section (rather than being limited to stool samples), and we can also perform types of perturbation experiments that would be unethical to carry out on human volunteers (addition of toxin or community destruction). 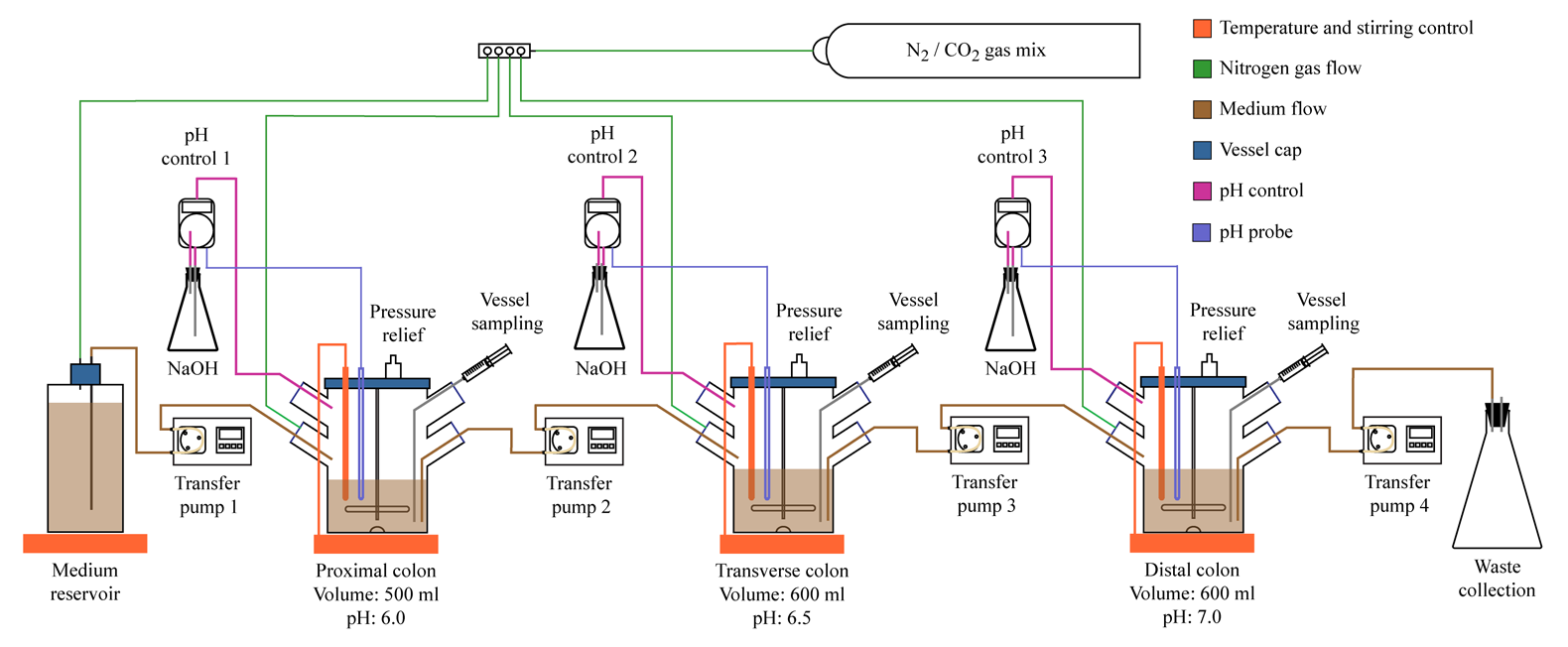
Figure. Schematic representation of in vitro simulator of human intestinal tract. |
|
Phylogenetic microarrays. In a recently completed project we have designed, developed, and validated a custom high-throughput phylogenetic Microbiota Array containing probes to 16S rRNA genes of 775 different microbial species (phylotypes) of human gut microbiota. The Microbiota Array is based on the Affymetrix, Inc. platform and contains sets of phylogenetic 16S rRNA gene probes (25-mer probes, 5-11 probe pairs in each set, each probe pair consists of perfect match and mismatch probes, each set interrogates a separate phylotype) allowing detection of 775 bacterial phylotypes of human intestinal microbiota. This design allows us to detect the presence and directly measure abundance level of all these species in each interrogated sample. Compared to the alternative technologies advantages of phylogenetic microarrays include (i) ability to measure each phylotype presence and abundance in all samples, (ii) fully quantitative nature of the acquired data allowing direct comparisons of levels of each phylotype among samples; and (iii) short processing and data acquisition times (only two days from sample to data using Microbiota Array). The designed microarray has been rigorously validated and used successfully in previous studies. We have shown that the microarray can be used not only to examine population structure of intestinal microbiota by profiling total genomic DNA isolated from fecal samples and biopsies, but it can also provide quantitative assessment of relative metabolic activities of individual community members through the interrogation of total community RNA. |

Figure. Microbiota Array. |
This Microbiota Array is now used to study the composition of human gut microbiota in a variety of recently completed and ongoing projects. The goal of many of these studies is to test whether gut microbial communities are changed in individuals diagnosed with different gastrointestinal diseases. If such differences are found, this would allow us to make putative associations between disease state and specific microbiota changes which can eventually lead to the development of novel drugs such as pro- and prebiotics. |
|
High throughput sequencing. High throughput sequencing (often also called next generation sequencing or NGS) is a novel approach that provides massive amounts of nucleotide sequence information in a single run. The main difference between a standard Sanger sequencing (which uses chain-termination method) and these new high-throughput methods is the parallelization of the sequencing reaction in NGS. Typically, millions of different nucleotide sequences can be read by NGS machines simultaneously. This allows researchers to obtain 100- and 1000-fold higher amount of data within the same amount of time as compared to Sanger method. The parallelization also lowers per-nucleotide sequence acquisition costs by several hundred folds. This technology has already been employed to study genome sequence variation, ancient DNA, DNA methylation, protein-DNA interactions, alternative splicing, small RNA populations, and mRNA transcription and regulation. NGS is especially well suited to the analysis of microbial communities. Since many such communities are composed of hundreds of different members, the overall community contains millions of different genes, which can only be profiled to reasonable depth by NGS. | 
Figure. Ion Torrent Personal Genome Machine. |
|
Recent research in our laboratory utilized three different NGS methods. 454-based pyrosequencing was used to confirm microarray results in the analysis of fecal microbiota in healthy and IBS children. Illumina HiSeq and MiSeq platforms were used to assess microbial community changes in patients undergoing fecal transplantation treatment for recurrent Clostridium difficile infection. And we employed Ion Torrent PGM machine to profile differences in gut communities of children consuming different diets. |
|
Fluorescent in situ hybridization. Fluorescent in situ hybridization (FISH) is a cell and DNA detection method that allows visualization and enumeration of DNA molecules based on the selective hybridization of specific fluorescently labeled oligonucleotide probes. In microbiological research, FISH can be utilized to selectively visualize specific species and cells in an environmental sample if we design probes uniquely targeting DNA in those cells. In a typical protocol, cell mixture is first fixed, the oligo probes are allowed to hybridize to DNA in cells, and after sample washing the probe signal is observed and measured under a fluorescent microscope. Because fluorescent in situ hybridization relies on direct visualization of cells, it serves as an excellent validation method for DNA quantification-based microarray and sequencing techniques. We have employed FISH to visualize Clostridia, Bacteroidia, Proteobacteria, and Bifidobacterium cells in fecal microbiota communities by employing FITC-labeled short oligonucleotide probes complementary to regions of 16S rRNA gene unique to each selected bacterial group. DAPI stain (binds to any DNA) was used to visualize all cells (see image). |
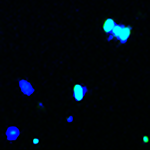
Figure. Visualization of Proteobacteria cells with FITC-labeled DNA probe. |
Anaerobic culturing. Majority of human gut microbiota members are obligate anaerobes, i.e., they use anaerobic fermentation pathways to generate energy from nutrients. These organisms cannot survive oxygen in their environment because they lack reactive oxygen species detoxifying enzymes such as catalase. In order to grow these organisms in the laboratory, special conditions are required in which all oxygen is removed from the incubator / environment. Several different methods can be utilized to cultivate anaerobic bacteria such as anaerobic chambers, boxes, and sacks, use of nitrogen and anaerobic atmosphere sparging, and addition of reducing compounds to the medium to bind oxygen. We use a combination of these approaches to cultivate individual members of human gut microbiota, which enables us to study each species independently and assess their metabolism, physiology, and interactions with other members of the community. |
 |
Metabolite profiling with proton NMR. In collaboration with Dr. Nicholas Reo (Department of Biochemistry and Molecular Biology, WSU) we have initiated an analysis of metabolic activity of gut microbial communities by using proton NMR measurements of fecal and lumenal extracts. Proton NMR enables us to detect and quantitatively measure levels of many different small organic compounds based on the magnetic properties of the hydrogen atoms bound to each carbon in the molecule. Similar to our microarray findings, stool samples from patients with a disease can be separated from healthy ones based on the metabolite levels in their stool samples. Some important metabolites such as short-chain fatty acids are often reduced in the lumen of people with GI disorders, and these metabolic changes can be associated with particular disease states. Since many of these metabolites are produced by resident microbiota, combining microarray and NMR analyses of the same samples will allow us to putatively link metabolite production / consumption and the presence and abundance of different bacterial members. |
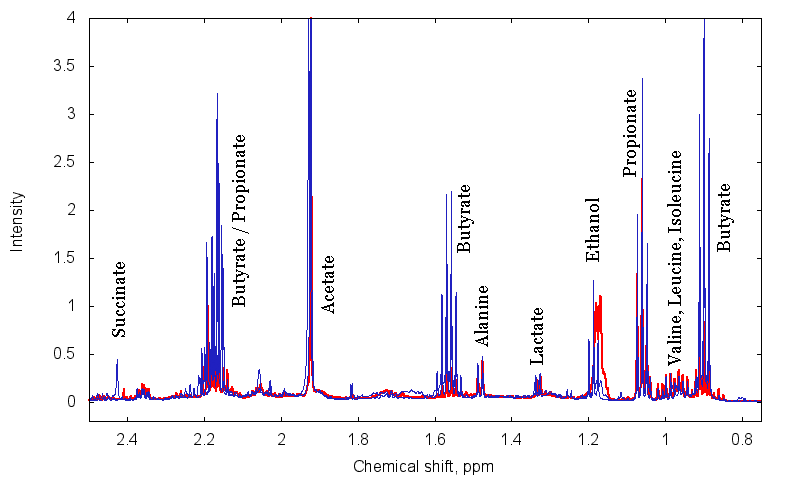
Figure. Aliphatic region of typical H1-NMR spectrum for two fecal extract samples. [use "view image" to expand] |
Metabolic modeling. Typical microbial communities encompass hundreds of different species and phylotypes, each filling a particular environmental niche. Many metabolic interactions are known and presumed to exist among these species, where end-products of metabolism of one species are released into the environment and are used as a source of nutrients and energy by another microbe (see image). However, due to the complexity of these communities, finding all such interactions experimentally is challenging. One alternative powerful approach to assess possible cross-species interactions is through mathematical modeling of each species metabolism and subsequent comparison of metabolic maps among different microbes. In this approach, we utilize known genome sequences of different community members to build metabolic maps for each of them based on the genome information. We then assess all possible metabolic interactions among community members. Such predictions can then be tested experimentally in a two or three-member community. |
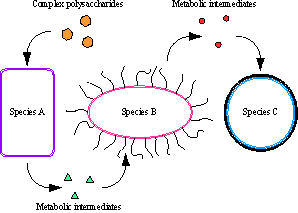
Figure. Schematic representation of metabolic interactions among microbiota. |
Computational biology and biostatistics. Many of the experimental approaches that we use in our research generate large amounts of data for many tens and hundreds of samples. An analysis of such large datasets requires special computational approaches that often have to be modified and tailored to each individual project. We therefore develop new and adapt existing biocomputational techniques to simplify the analysis of such data and produce revealing visualizations of the major findings. Among the techniques that we utilize are ordination and classification algorithms such as principal components analysis (PCA), phylogenetically based principal coordinates analysis (PCoA), and orthogonal projection to latent structures discriminant analysis (OPLS-DA), correlation matrix calculations, ecological diversity analyses, data clustering, and compositional analysis. |
 Figure. Sample of Matlab code that runs PCA ordination analysis [use "view image" to expand]. |
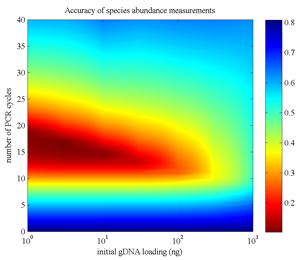 Figure. Modeling the accuracy of microbial abundance measurements of microbial as a function of PCR amplification parameters (lower is better) [Paliy and Foy Bioinf 2011]. |
|
We also employ a variety of biostatistical approaches to test the statistical significance of the observed differences among samples and between sample groups. In addition to classical ANOVA and regression analyses, these methods include generalized linear models, permutation analyses, rarefaction calculations, and multiple hypothesis testing adjustments. |
||